
Genomic analyses of the
transcription elongation factor Spt6
To access the information encoded within a gene, the DNA sequence of the gene must be transcribed to produce RNA. The process of transcription is carried out by a protein complex called RNA polymerase, and occurs in three stages of initiation, elongation, and termination. During each of these stages, RNA polymerase is associated with factors which modulate its activity and carry out co-transcriptional processes.
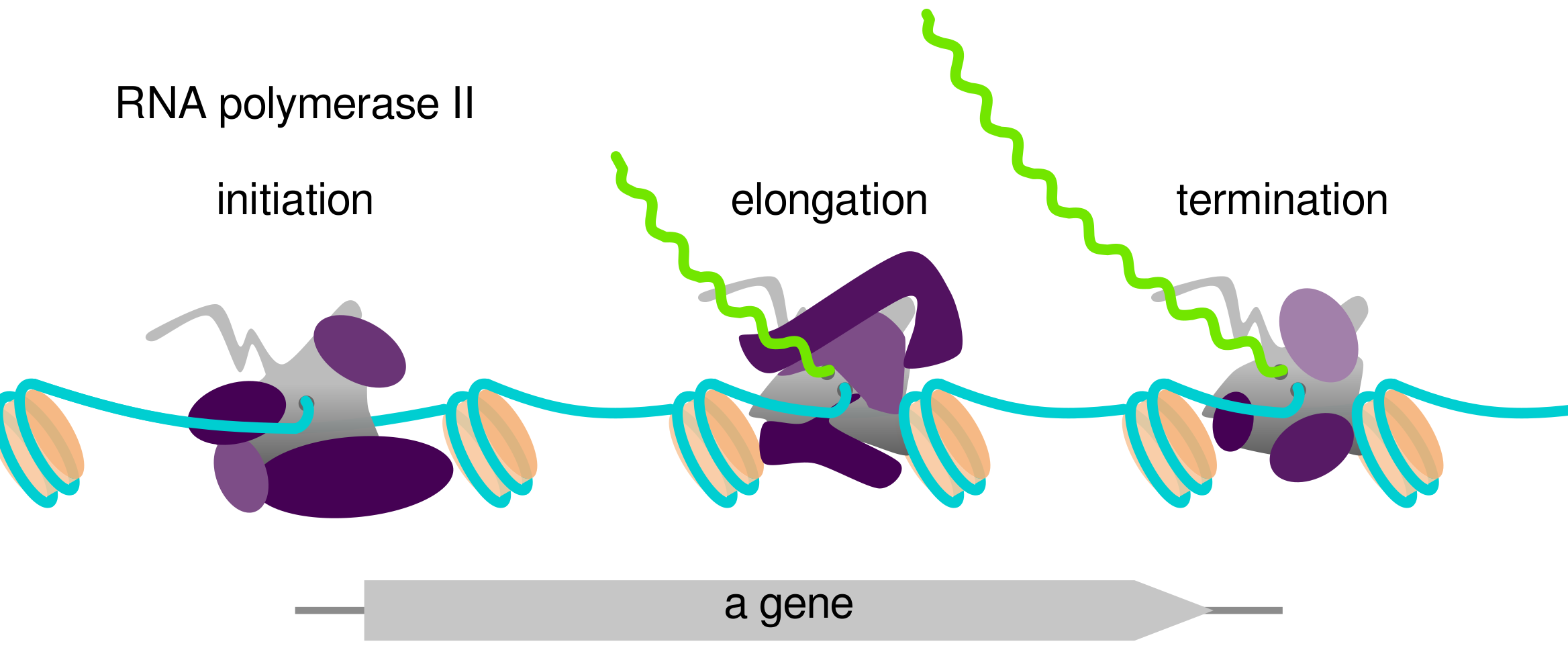
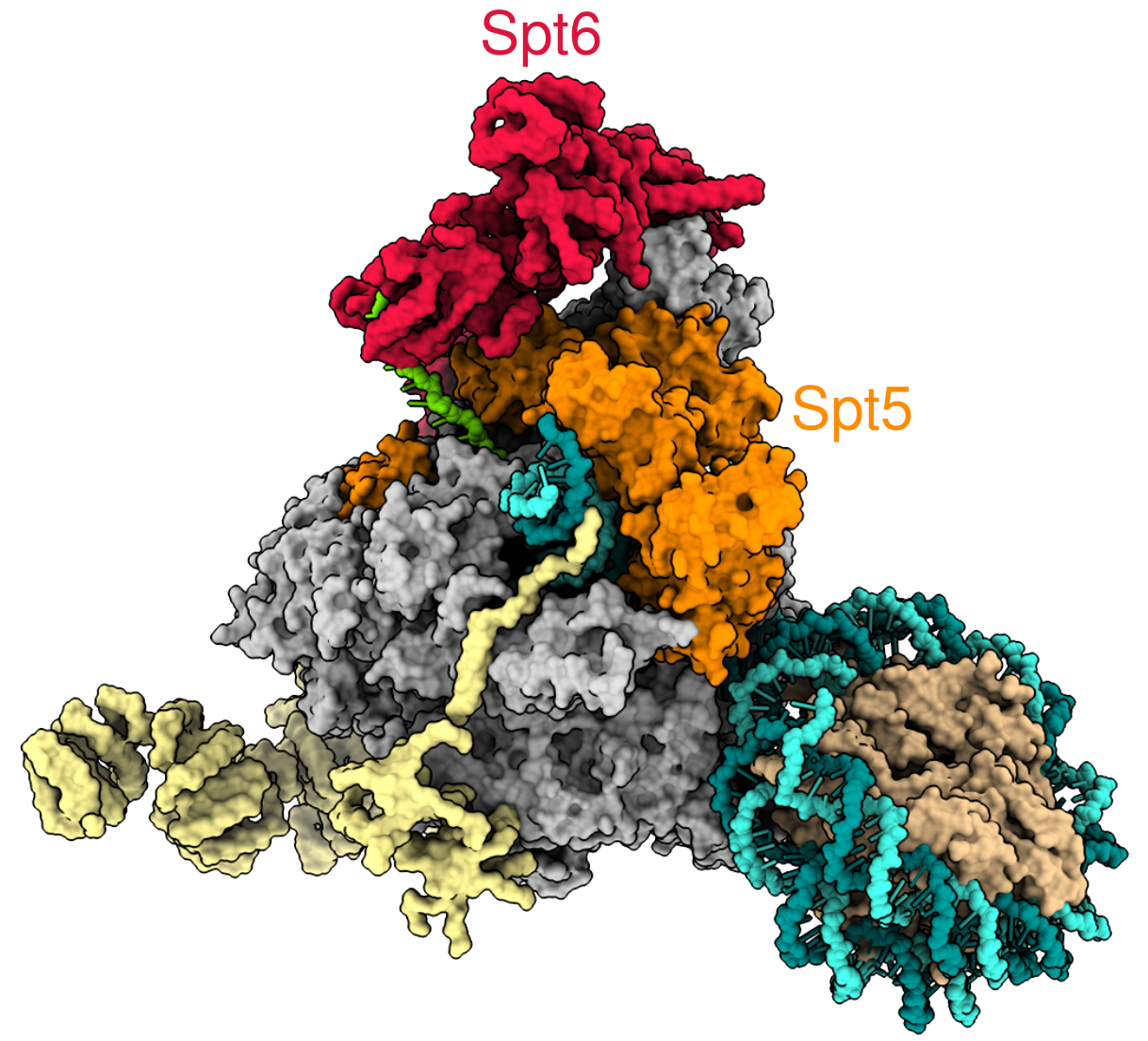
As a graduate student in the Fred Winston lab, I worked on a project studying a protein called Spt6, one of the factors which associates with RNA polymerase during eukaryotic transcription elongation. One of the major functions of Spt6 is to be a histone chaperone, assisting with the re-assembly of histones and DNA into nucleosomes. To study Spt6, my collaborators in the lab generated a number of genomic datasets comparing wild-type yeast cells to yeast cells with a mutation in Spt6, called spt6-1004. We performed these experiments using yeast because the process of transcription is very similar between yeast and humans, and yeast is extremely convenient to work with. One phenotype of spt6-1004 cells we were particularly interested in studying is the phenomenon of intragenic transcription, unusual transcription which starts in the middle of a gene.
The genomic assays my collaborators performed to study Spt6 are listed in the following table:
| assay | description |
|---|---|
| TSS-seq | Reports the positions of the 5′-ends of transcripts, i.e. transcription start sites (TSSs) |
| ChIP-nexus | A high-resolution ChIP-seq technique which reports the occupancy of a protein of interest on the genome |
| NET-seq | Reports the locations of actively transcribing RNA polymerase |
| MNase-seq | Measures nucleosome occupancy and positioning over the genome |
To analyze the data produced by these assays in a reproducible manner, I developed analysis pipelines using the Snakemake workflow management system. The pipelines are maintained at the Winston Lab github page. A few examples of figures I produced using these pipelines are below.
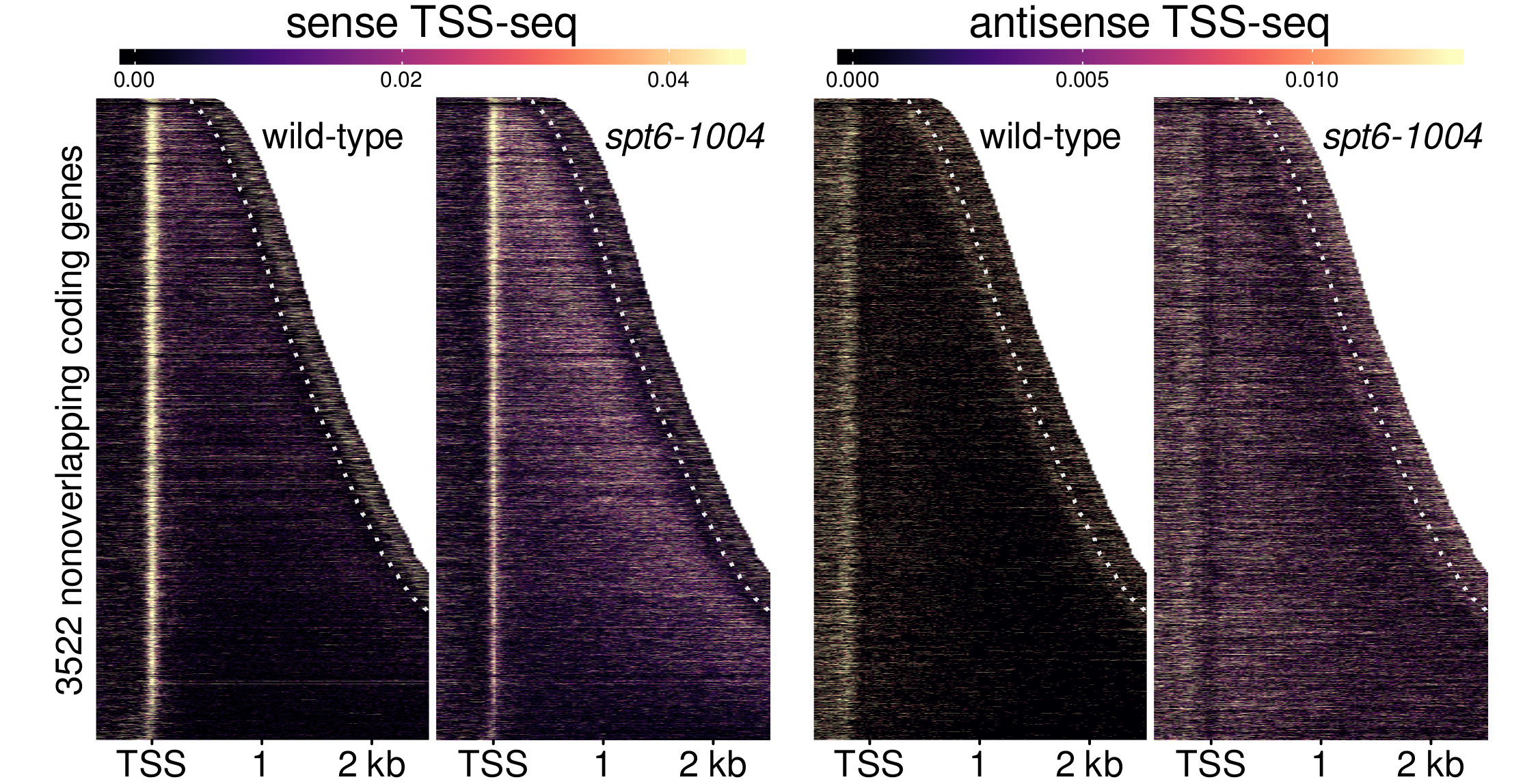
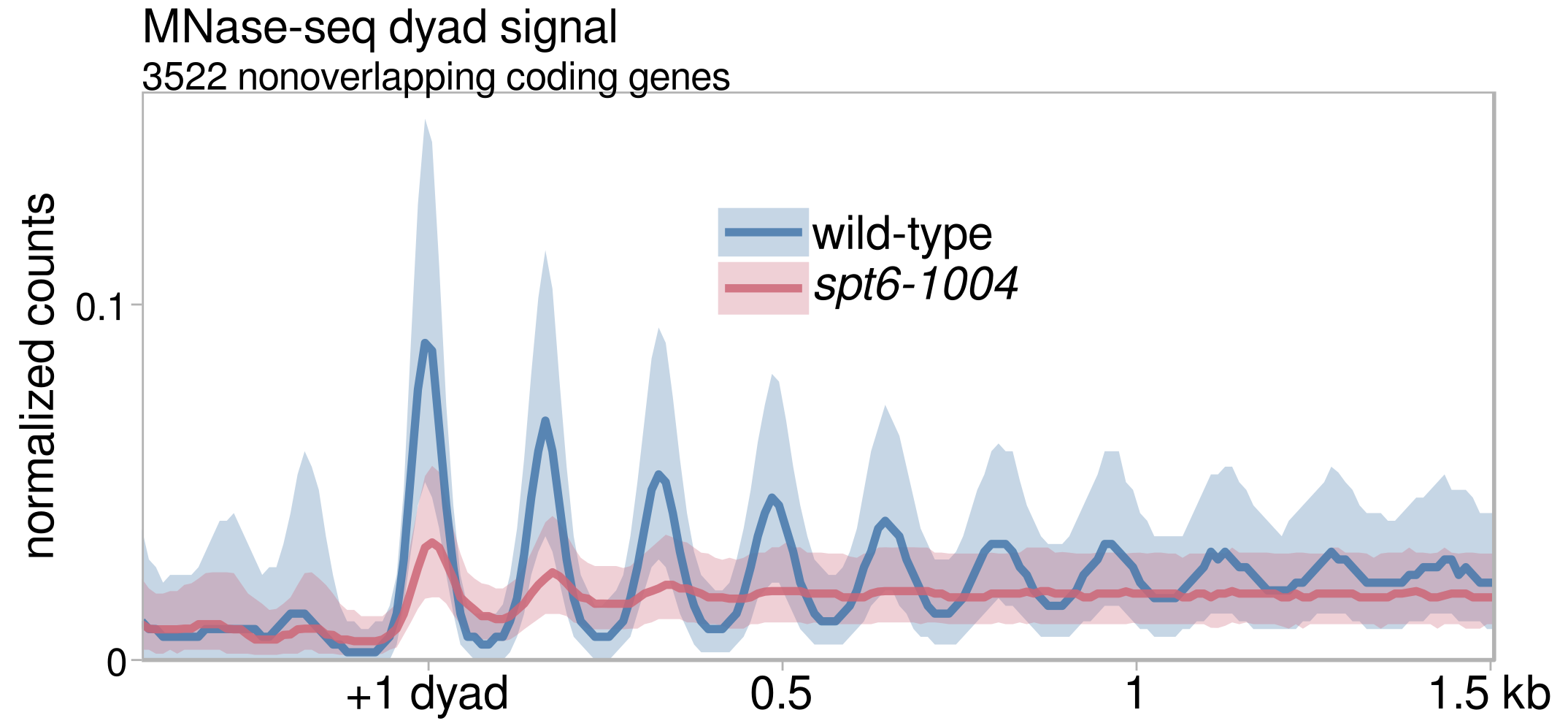
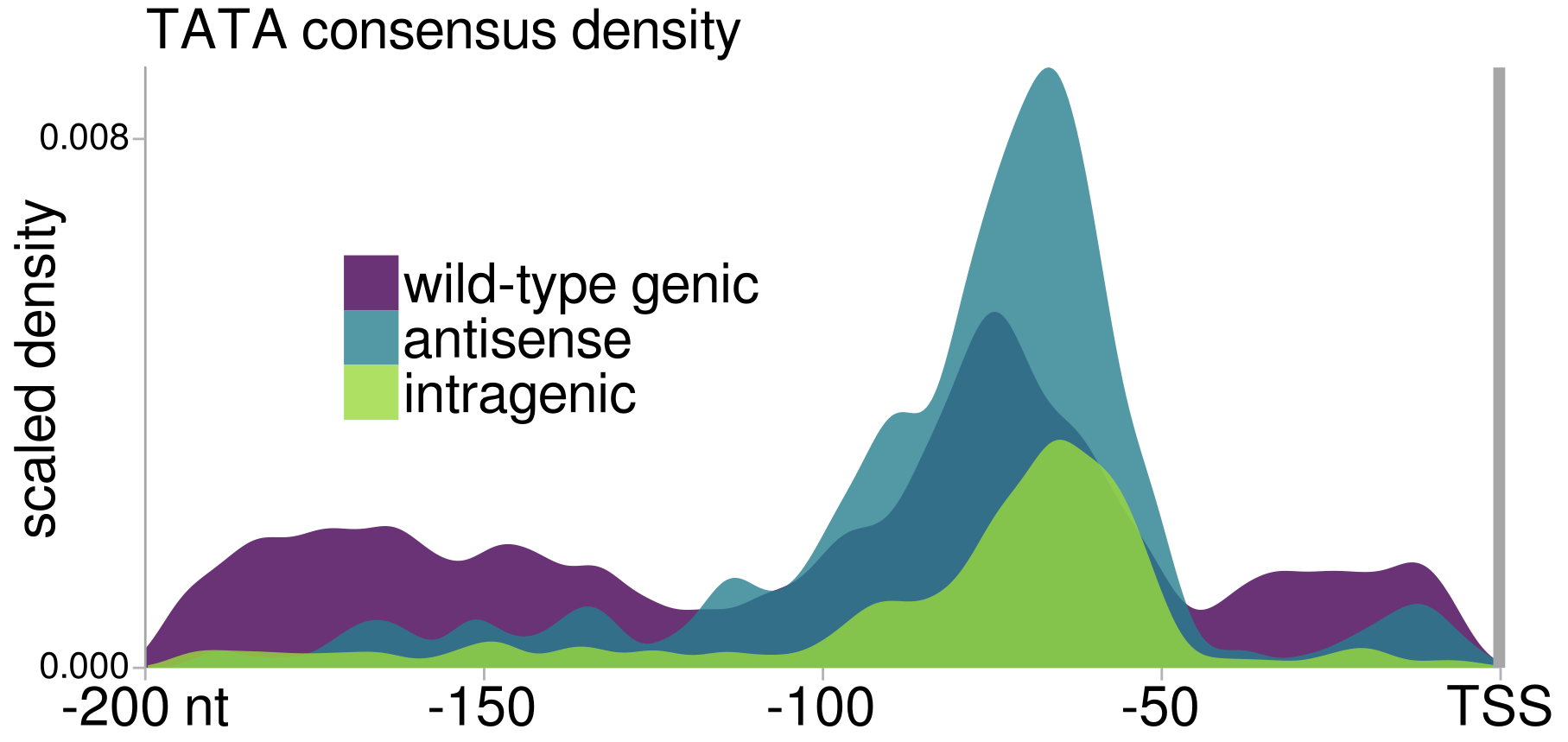
Our results were published in Molecular Cell, with the following major conclusions:
- Using TSS-seq, we catalog the full extent of intragenic transcription in spt6-1004, finding thousands of upregulated intragenic and antisense transcripts.
- Using ChIP-nexus of TFIIB, we show that TFIIB binding is widespread over the genome in spt6-1004, and that new transcription initiation explains most intragenic transcripts in spt6-1004.
- Using MNase-seq, we observe a global depletion and disordering of nucleosomes in spt6-1004.
- We find that intragenic promoters induced in spt6-1004 have some sequence features of canonical genic promoters.
- By both TSS-seq and TFIIB ChIP-nexus, we observe an unexpected decrease in genic transcription in spt6-1004.
- Altogether, we propose that the reduction in nucleosome protection of the genome in spt6-1004 makes promoter-like sequences within genes accessible to the transcription initiation machinery, causing a redistribution of transcription initiation factors away from genic promoters and towards intragenic promoters.
Upon publication, I made the raw data and all data analyses available on Zenodo, allowing anyone interested to go all the way from raw data to the figures in the publication.
Overall, our studies highlight the importance of nucleosomes in restricting access to the genome, and the role of histone chaperones like Spt6 in maintaining nucleosomes in their normal state.